
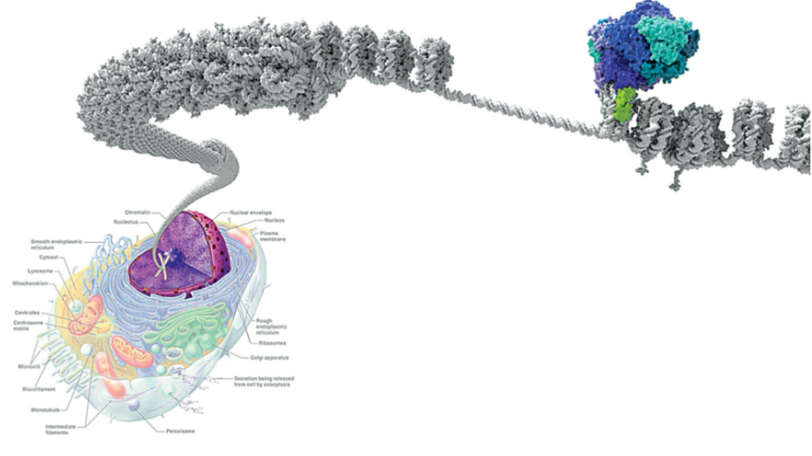
染色质重塑是基因表达动态调控的重要机制之一,主要由不同的蛋白质/蛋白质复合物完成。SWI/SNF复合物是其中之一,转录调控因子将SWI/SNF复合物募集到DNA区域,沿DNA移动并通过变构核小体修改DNA的可访问性或可及性(重塑染色质),通过转录调节因子与暴露的DNA的结合实现基因表达调控(抑制或者激活)。SWI/SNF亚基基因在20%以上的恶性肿瘤中发生突变,其亚基的突变会给癌症带来脆弱性,为新药研发提供方向[1]。
什么是SWI/SNF复合物?
DNA 包裹在组蛋白周围形成核小体,这些核小体又盘绕成致密的染色质(图1灰色)。染色质处于紧缩状态下无法读取DNA信息。因此,一种精细的细胞机制与转录因子协同工作以动员核小体从而控制基因表达,这一过程称为染色质重塑。染色质重塑复合物SWI/SNF也称为 BRG1/BRM (BAF) 复合物(图1蓝色)通过水解ATP获得能量移动核小体并去除组蛋白核心解开DNA,开放状态下DNA能被转录。SWI/SNF复合物不仅与启动子结合,还与其他调控区域(如增强子和DNA复制起始区)紧密结合。此外,SWI/SNF可结合/共沉淀许多蛋白质,在细胞周期等过程中发挥作用。这表明SWI/SNF复合物的功能比单纯的转录调控更为广泛[2]。
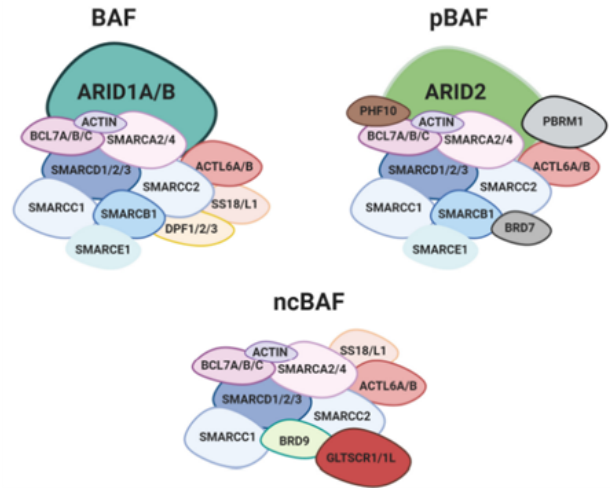
SWI/SNF复合物亚基组成
SWI/SNF 复合物是复杂的大分子组装体,由许多不同和可变的亚基组成。SWI/SNF复合物分成典型BAF复合物(cBAF)、多溴相关BAF复合物(pBAF)、一种新发现的复合物(ncBAF)三个大类,每一类具有不同的亚单位结构和功能,三种类型的BAF都包含核心亚基SMARCC1、SMARCC2和ATP酶(SMARCA4或SMARCA2),协同调节染色质的状态。此外三种类型既有共享亚基也包括特异性亚基,其中ncBAF包含特异性亚基(GLTSCR1 / 1L和BRD9)[3]。SWI/SNF 复合物中任何一个亚基的缺陷都可能会产生严重后果。在本文中我们将重点关注 SWI/SNF复合物亚基突变致癌的作用。
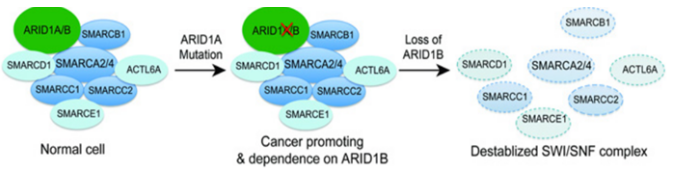
SWI/SNF复合物亚基突变与肿瘤
癌症基因测序揭示,大约25%的癌症存在一个或多个 SWI/SNF亚基基因异常,总共至少有九种不同的SWI/SNF亚基被鉴定为在各种癌症中反复发生突变。那SWI/SNF复合物亚基突变导致癌症发展的机制是什么?影响 SWI/SNF 亚基功能的突变是否会给这些癌症带来脆弱性?接下来将列举几种主要的亚基突变与癌症发生机制,以及相应的抗肿瘤治疗策略。
靶向SWI/SNF复合物ARID1A和 ARID1B
目前研究发现编码 SWI/SNF 亚基的基因的突变通常会对编码其他 SWI/SNF 亚基的基因产生特定的依赖性,亚基突变不会完全丧失SWI/SNF 功能,而是依赖于SWI/SNF 复合物同源亚基的活性导致异常细胞功能。ARID1A 突变细胞系特别依赖于其旁系同源物ARID1B,这是最早阐明旁系同源物依赖性的研究之一。ARID1A被发现在近 50% 的卵巢透明细胞癌 (OCCC) 和卵巢子宫内膜样癌 (OEC) 中发生突变。SWI/SNF 复合物中的 ARID1B是ARID1A 互斥同源物,在肿瘤中ARID1A的突变导致SWI/SNF 复合物产生了对 ARID1B 的依赖,研究发现在ARID1A突变细胞中抑制ARID1B 会使 SWI/SNF 复合物不稳定并影响细胞增殖[3]。
SMARCA4和SMARCA2同样,已显示SMARCA4突变细胞系对其旁系同源物SMARCA2的依赖性。SMARCA2和SMARCA4为SWI/SNF的ATP水解酶,负责染色质的重塑和修复,SMARCA4是继ARID1A之后恶性肿瘤中第二频繁突变的SWI/SNF基因,其作用比SMARCA2更重要。SMARCA4在很多缺乏可靶向癌基因的癌症中突变,包括10-20%的非小细胞肺癌、100%的小细胞卵巢癌、28%的皮肤癌、16%的胶质瘤,以及14%的结肠癌。SMARCA4突变癌细胞更依赖SMARCA2,因此选择性SMARCA2抑制剂或降解剂可以诱导SMARCA4缺陷型癌细胞死亡。这些发现表明了一种机制,即 SWI/SNF 亚基的丢失由旁系同源物部分补偿,使旁系同源物具有一种特殊的脆弱性[2]。

SMARCB1和BRD9这种复杂的内部依赖性不限于旁系同源亚基,目前已显示SMARCB1突变细胞系对非旁系 SWI/SNF 亚基 BRD9的依赖性增加。滑膜肉瘤 (SS) 是一种罕见的间叶来源恶性肿瘤,约占所有软组织肉瘤 (STS) 的8%至10%,SS几乎可以发生在任何部位,SS具有高转移性。融合蛋白SS18-SSX在100%的滑膜肉瘤病例中发现,SS18-SSX融合蛋白取代SWI/SNF中正常的SS18亚基。可能由于融合蛋白体积较大,SMARCB1亚基从滑膜肉瘤SWI/SNF复合物中被置换,SMARCB1被排出及降解导致功能丧失。由于破坏两个含有 SMARCB1 的亚基的SWI/SNF 复合物类型(cBAF 和 pBAF),进而增加对第三个亚家族(ncBAF)的依赖。研究发现ncBAF的特异性BRD9亚基的化学和生物消耗迅速减弱SS细胞增殖,这些发现揭示了 ncBAF 复合物独特的染色质靶向和功能,并提出了新的癌症特异性治疗靶点。
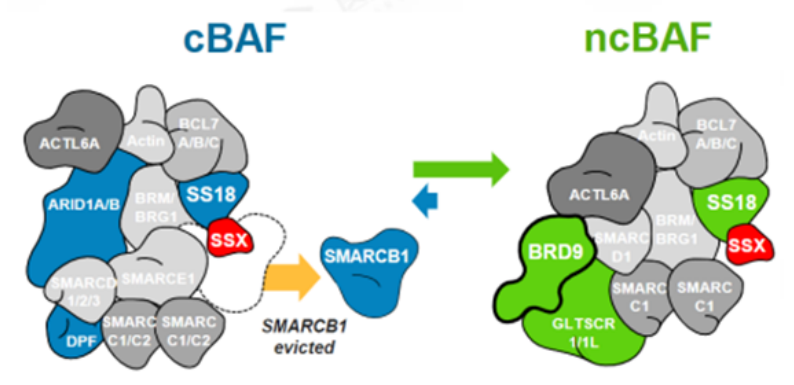
图5. SS18-SSX融合蛋白替换SMARCB1示意图(图片来源C4 Therapeutics官网)靶向非SWI/SNF 复合物SMARCB1和EZH2研究表明 SWI/SNF 复合物和与其他染色质修饰物家族Polycomb 阻遏物复合物(PRCs)具有相反的基因调节功能。在正常细胞内,PRC2通过其酶亚基(EZH2) 催化维持H3K27三甲基化 (H3K27me3) 标记,促进染色质凝集,从而抑制转录。SWI/SNF 复合物定位于H3K27ac位点,与转录因子合作建立开放的染色质状态,促进转录。SWI/SNF复合物对PRC2(含有EHZ2亚基)起到抑制作用,促进PRC2的靶基因的表达,促进转录的进行[2]。
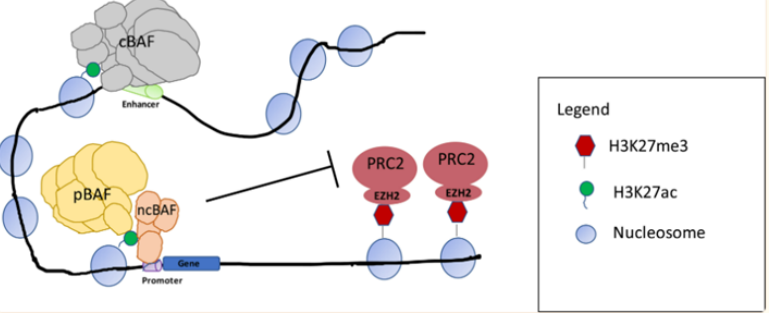
复合物与PCR2定位[2]SMARCB1(INI1)的缺失在90%以上的上皮样肉瘤(ES)病人中发生,且成为ES的一个典型的诊断特点。在ES中 SMARCB1发生突变或缺失时导致EZH2活性增强,H3K27me3水平升高,造成组蛋白和DNA紧密结合,很多抑癌基因的表达受到抑制,而hedgehog、MYC等信号通路基因等上调,促使细胞增殖,形成肿瘤。研究发现在ES中,使用EZH2抑制剂特异性作用于靶点EZH2时,降低EZH2活性,最终染色质的“捆绑和束缚”得到释放,基因得以正常表达,达到杀死肿瘤的效果。EZH2抑制剂Tazemetostat在2020年1月获得FDA批准,用于治疗16岁及以上、不符合完全切除条件的转移性或局部晚期ES儿童和成人患者。
ARID1A与ATR研究发现SWI/SNF 复合物还与 DNA 损伤修复 (DDR)的几种机制有关。不同的 SWI/SNF 家族成员已被证明在 DDR 中具有不同的作用,从重塑DNA 损伤位点周围的染色质结构到直接招募 DDR所需的蛋白质。cBAF和pBAF复合物涉及非同源末端连接 (NHEJ) 和同源重组 (HR)修复过程。ARID1A 通过与 ATR 的相互作用被招募到DNA双链断裂(DSB)处,为了应对 DNA 损伤,ARID1A 促进 DNA DSB 末端加工以生成 RPA 包被的单链 DNA (ssDNA),并维持 ATR 激活以响应 DSB。ARID1A 的缺失导致检查点激活和DSB 修复受损,这使细胞对 DSB 诱导治疗敏感,例如PARP抑制剂,ATR抑制剂[6]。临床前数据证明了 ARID1A 缺失与 VX-970(ATR 的抑制剂)之间的协同作用,以及 PARP 抑制剂与放射治疗在 ARID1A 突变肿瘤中的功效。PARP 或 ATR 抑制剂目前正在几项ARID1A 突变癌症患者的试验中被评估[2]。
2022AACR公布的SWI/SNF复合物亚基突变治疗新策略
BRD9靶向抑制剂
CFT8634根据上文的机制在滑膜肉瘤病例发现100%SS18-SSX融合蛋白,针对SS18-SSX融合蛋白本身很难开发药物,根据SS18-SSX融合导致SMARCB1脱离cBAF、pBAF复合物,使得染色质重塑复合物更依赖ncBAF, 那么靶向ncBAF复合物的特异性亚基BRD9就有可能抑制SMARCB1缺失肿瘤[5]。C4 Therapeutics公司的CFT8634是一款双特异性分子,它能够将BRD9和E3泛素连接酶CRBN连接在一起,在BRD9蛋白上添加泛素修饰,导致BRD9的特异性降解。传统的小分子抑制剂仅仅限制了乙酰赖氨酸溴化酶的阅读功能,而降解技术能够最大化破坏ncBAF复合物的致癌活性。在滑膜肉瘤的临床前模型中,CFT8634表现出持续降低肿瘤体积的效果。这款在研疗法已经获得美国FDA授予治疗滑膜肉瘤的孤儿药资格,其IND申请在今年2月底获得许可,预计在今年上半年开始针对滑膜肉瘤患者的I期临床试验。
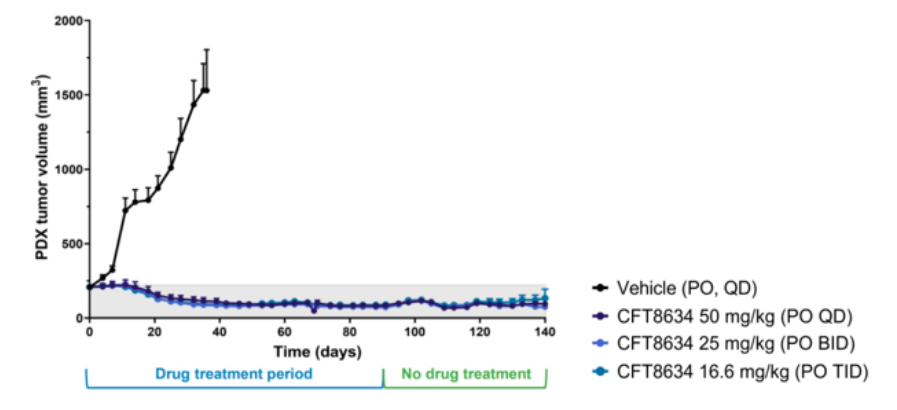
图7. CFT8634临床前肿瘤抑制效果(图片来源:C4 Therapeutics官网)Foghorn公司研发的FHD-609也是BRD9的选择性蛋白质降解剂,在临床前研究中,FHD-609已被证明可以选择性地降解BRD9,利用与SS18-SSX易位的合成致死关系杀死癌细胞,目前FHD-609是第一个进入临床的BRD9降解剂。
SMARCA2选择性抑制剂PRT3789SMARCA2和SMARCA4 都是SWI/SNF复合物的ATP酶,负责染色质的重塑和修复。SMARCA4突变癌细胞依赖旁系SMARCA2,因此选择性SMARCA2抑制剂或降解剂可以诱导SMARCA4缺陷型癌细胞死亡。在SMARCA4突变肺癌细胞系中的全基因组CRISPR筛选表明,MCL1的丢失可以使SMARCA4突变肺癌细胞对SMARCA2降解敏感,Prelude Therapeutics公司的SMARCA2降解剂 PRT3789 联用MCL1抑制剂PRT1419在SMARCA4突变肺癌模型中产生协同作用,并且不影响SMARCA4野生型细胞。PRT3789对SMARCA2选择性强(超SMARCA4 20倍),体内外模型耐受良好,Prelude预计2022年年底申报临床。
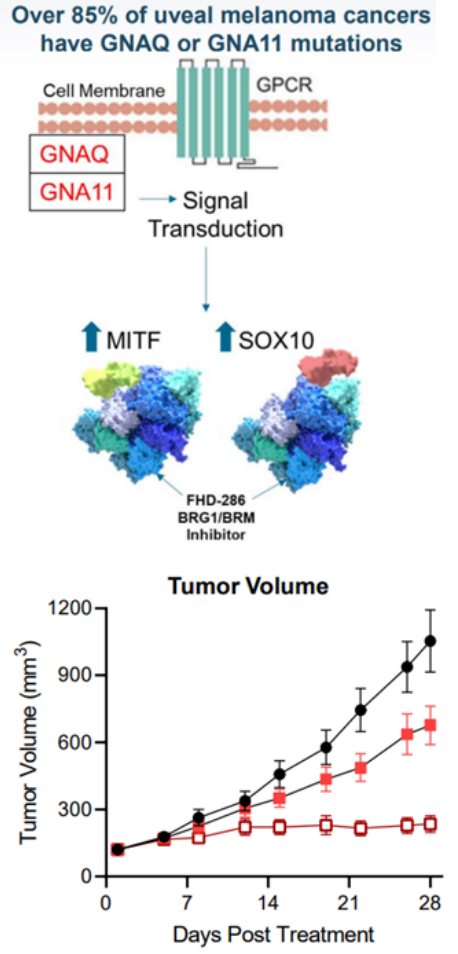
SMARCA2/SMARCA4双抑制剂FHD-286Foghorn今年AACR披露首个进入临床的SMARCA2/SMARCA4双抑制剂 FHD-286临床前数据。FHD-286是Foghorn公司一款BAF复合物口服抑制剂,用于治疗AML和葡萄膜黑色素瘤(UM)。FHD-286通过抑制BAF复合体中的ATP酶成分SMARCA4和SMARCA2,抑制BAF的功能。FHD-286抑制SMARCA2/SMARCA4影响SPI1转录水平及其下游转录程序,从而影响细胞增殖和细胞存活。SPI1是一种 ETS 家族转录因子,在造血发育和分化中起关键作用,SPI1表达调节与急性髓性白血病 (AML) 的肿瘤发生有关。临床前证据显示FHD-286在AML患者样品中广泛有效,低纳摩尔剂量具有促进细胞分化的作用,细胞杀伤作用与标准疗法相当,剂量依赖性抑制肿瘤生长(图8)。目前FHD-286已开展针对AML/MDS的I期临床试验。

在85%葡萄膜黑色素瘤(UM)中存在GNAQ / GNA11突变,其转录因子MITF和SOX10过表达并与BAF复合物过度相互作用,FHD-286抑制SMARCA2/SMARCA4,导致SOX10 和 MITF 转录因子结合位点的可及性丧失,抑制依赖SOX10和MITF的基因(GNAQ / GNA11)表达,临床前数据显示FHD-286剂量依赖性抑制肿瘤生长(图9)。目前FHD-286已开展针对UM的I期临床试验。
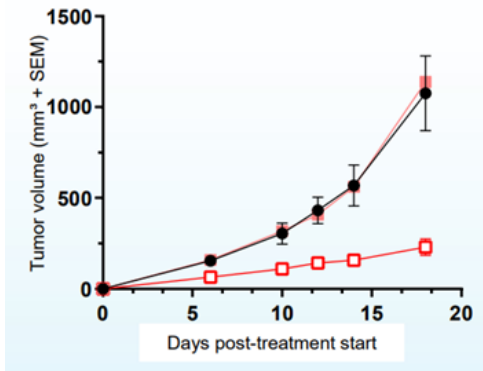
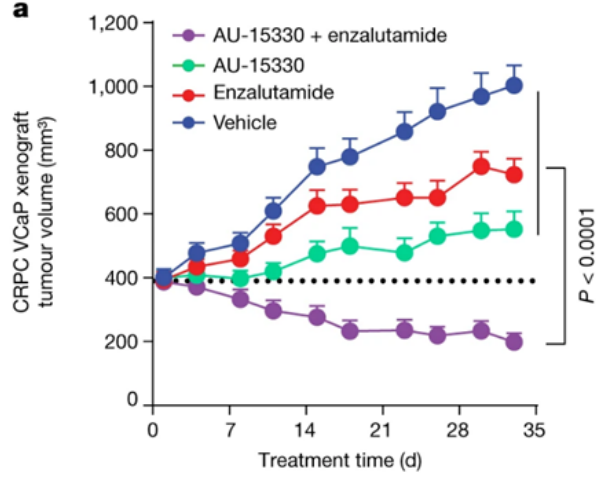
AU-15330SWI/SNF复合物的组分在一些癌症中发生了突变,但在前列腺癌中很少发生。相比于SWI/SNF复合物的亚基发生突变的癌症,由雄激素受体(AR)或FOXA1驱动的前列腺癌对SWI/SNF降解剂更敏感。AU-15330是Aurigene 公司的一款SWI/SNF ATP酶亚基(SMARCA2和SMARCA4)的蛋白水解靶向嵌合体降解剂。主要的作用机制是降解SMARCA2和SMARCA4,阻止对染色质的访问,压缩AR、FOXA1、ERG 和 MYC 的核心增强子周围的染色质,转录因子被阻止与驱动癌症的增强子结合,从而减弱促癌转录程序[9]。AU-15330 在前列腺癌异种移植模型中可有效抑制肿瘤生长,并与 AR 拮抗剂 enzalutamide 协同作用(图10)。
相关新闻
◎版权作品,未经今日健康网书面授权,严禁转载,违者将被追究法律责任。
Copyright 2015-2018. 今日健康网 www.jinrijiankang.org All rights reserved.
违法和不良信息举报邮箱:jubao@jinrijiankang.org 执行主编:为民
未经过本站允许,请勿将本站内容传播或复制



